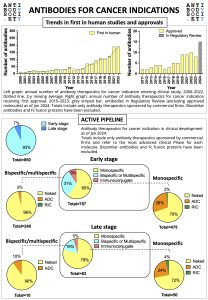
For World Cancer Day 2024, The Antibody Society has prepared a snapshot of the clinical development of therapeutic antibodies for cancer indication.
The infographic gives an overview on the trends in first in human studies and approvals, as well as on the active early and late stage pipelines (as of January 2024).