
Antibody Engineering & Therapeutics, held in December 2021, offered many opportunities to hear exciting and informative presentations by experts in the field. We are pleased to present here a summary of a plenary lecture by Prof. Charlotte Deane (University of Oxford), kindly written by Dr. Czeslaw Radziejewski.
Application of Machine Learning and Informatics in Antibody and Protein Research
Charlotte Deane, Professor of Structural Bioinformatics, Department of Statistics, University of Oxford
Machine learning relies heavily on the availability of large databases. Three databases for antibody research were developed in Prof. Dean’s lab: OAS (Observed Antibody Space),[1] SAbDab (Structural Antibody Database),[2] and Thera-SAbDab (database of immunotherapeutic variable domain sequences). OAS contains about 2 billion redundant antibody sequences across diverse immune states, organisms, and individuals. SAbDab is a fully automated self-updating collection of publicly available antibody structure data. It contains 5650 structures, but about 1000 truly non-redundant structures, 4213 antigen-antibody complexes and 890 structures of nanobodies. Thera-SabDob contains 696 structures as of October 2021. In addition, the lab has a CoV-AbDab database that contains sequences and structures for coronavirus antibodies for SARS-CoV-2, SARS-CoV-1 and MERS-CoV. This database contains about 5000 data points. The lab developed the SAbPred suite of tools for antibody prediction, comprising AntibodyBuilder, SPHINX, SCALOP, PEARS, ANARCI, ABangle, Hu-mAb SAAB+,TAP, Epitope Profiling SPACE and Ab-Ligaty. SCALOP, ABodybuilder, SPHINX are designed for building antibody models. ABlooper tool builds complementary-determining region (CDR) structures. ABangle is a tool for calculating and analyzing the VH-VL orientation in antibodies. TAP (Therapeutic Antibody Profiler) considers the drug-like properties of therapeutic antibodies.[3] It evaluates variable domains in antibody of interest using five developability criteria derived from post clinical Phase 1 antibody therapeutics. Epitope Profiling-SPACE and Paratyping Ab-Ligity can used to determine if two antibodies with divergent sequences can bind to the same epitope.[4] ANARCI is a tool for annotating antibody sequences and Hu-Mab is a computational tool for antibody humanization. Dlab is a deep learning method for virtual screening of antibody sequences that can bind specific antigens.
Professor Deane provided examples of using some of her computational tools. Antibody humanization is currently inefficient, as it is carried out experimentally in a largely trial and error process. Applying machine learning to an edited OAS database (with redundancies removed) led to classifiers that could distinguish between human and non-human antibody variable domain sequences. These classifiers were used to create the computational humanization tool Hu-mAb. Available sequences of therapeutic antibodies from different stages of development were subjected to Hu-mAb analysis. The high Hu-mAb scores correlated with low observed immunogenicity of an antibody and low scores correlated with higher observed immunogenicity. Twenty-five experimentally humanized antibody sequences for which rodent or rabbit precursor sequences were available were assessed by Hu-mAb. Most of the mutations that Hu-mAb generated were either the same or chemically similar for VH (77% and 85%, respectively) and for VL (59% and 58%, respectively). Hu-mAb suggested overall fewer mutations and fewer mutations to VH-VL interface than the experimental approach, therefore such humanized antibodies would more likely have preserved structure and function.
The Therapeutic Antibody Profiler evaluates properties thought to determine antibody developability, including CDRH3 or total CDR length; patches of surface hydrophobicity across CDR vicinity; patches of positive charges and negative charges across CDR vicinity; and structural Fv charge symmetry. These properties are related to aggregation, viscosity, poor expression and polyspecificity of antibody molecules.[5] TAP was applied in a study that used 137 post Phase 1 therapeutic models,14000 representative Human Antibody Models and 2 datasets of MedImmune Developability Failures. The study revealed that therapeutic antibodies tend to have shorter CDRH3 and smaller hydrophobic patches than natural ones. However, positive and negative patches of natural and therapeutic antibodies have similar profiles and Fv charge symmetry is also very similar. Both therapeutic and natural antibodies have an aversion to strongly oppositely charged VH and VL chains.
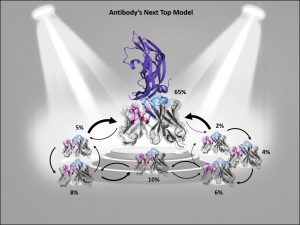
ABlooper [6] uses similar architecture as AlphaFold. It predicts structures of all six CDR loops and estimates the accuracy of prediction. The root-mean-square deviation from AlphaFold2 for CDRH3 prediction (2.87A) were comparable with ABlooper (2.49 A). Unlike AlphaFold2, ABlooper generates a series of predicted structures from which a prediction of accuracy can be estimated. If the predicted structures are widely divergent, then the quality of prediction is low. ABlooper is also much faster than other deep learning methods such as AlphaFold (100 structures predicted in 5 second vs one structure in 20 min). All tools are available freely for academic institutions.
- Olsen TH, Boyles F, Deane CM. Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci. 2022 Jan;31(1):141-146. doi: 10.1002/pro.4205.
- Schneider C, Raybould MIJ, Deane CM. SAbDab in the age of biotherapeutics: updates including SAbDab-nano, the nanobody structure tracker. Nucleic Acids Res. 2022 Jan 7;50(D1):D1368-D1372. doi: 10.1093/nar/gkab1050.
- Raybould MIJ, Deane CM. The Therapeutic Antibody Profiler for Computational Developability Assessment. Methods Mol Biol. 2022;2313:115-125. doi: 10.1007/978-1-0716-1450-1_5.
- Wong et al. Ab-Ligity: identifying sequence-dissimilar antibodies that bind to the same epitope. MAbs 2021. DOI: 10.1080/19420862.2021.1873478.
- Khetan et al. Current advances in biopharmaceutical informatics: guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. MAbs 2022. DOI: 10.1080/19420862.2021.2020082.
- Abanades B, Georges G, Bujotzek A, Deane CM. ABlooper: Fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinformatics. 2022 Jan 31:btac016. doi: 10.1093/bioinformatics/btac016.
 The
The