On June 15, 2023, the U.S. Food and Drug Administration (FDA) approved Columvi® (glofitamab-gxbm) for the treatment of adult patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) not otherwise specified or large B-cell lymphoma (LBCL) arising from follicular lymphoma, after two or more lines of systemic therapy. This indication is approved under accelerated approval based on response rate and durability of response in the Phase 1/2 NP30179 study. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
On June 15, 2023, the U.S. Food and Drug Administration (FDA) approved Columvi® (glofitamab-gxbm) for the treatment of adult patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) not otherwise specified or large B-cell lymphoma (LBCL) arising from follicular lymphoma, after two or more lines of systemic therapy. This indication is approved under accelerated approval based on response rate and durability of response in the Phase 1/2 NP30179 study. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
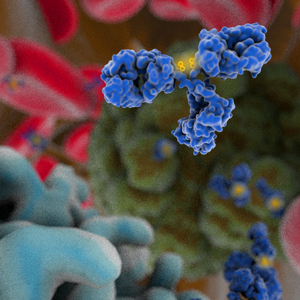
Glofitamab (RO7082859, CD20-TCB, RG6026) is a full-length IgG1λ/ҡ bispecific T cell-redirecting antibody targeting CD20 on malignant B cells and CD3 on T cells. This bispecific antibody was developed by Roche using the 2:1 CrossMab technology, characterized by 3 antigen-binding fragment (Fab) arms enabling monovalent binding to CD3ɛ and bivalent binding to CD20, with the second CD20 arm fused to the CD3ɛ-binding arms via a flexible linker. Glofitamab also features a heterodimeric Fc region engineered with PG LALA mutations to abolish binding to FcɣRs and C1q.
The FDA accelerated approval is based on positive results from the Phase 1/2 NP30179 study of Columvi given as a fixed course for 8.5 months in 132 patients with DLBCL who had relapsed or were refractory to prior therapies, including 30% who had received prior CAR T-cell therapy. Additionally, 83% were refractory to their most recent therapy. Results showed patients treated with fixed-duration Columvi achieved durable remission, with 56% of patients achieving an overall response (OR; 74/132 [95% confidence interval (CI): 47-65]) and 43% of patients achieving a complete response (CR; 57/132 [95% CI: 35-52]). Over two-thirds of those who responded continued to respond for at least nine months (68.5% [95% CI: 56.7-80.3]). The median duration of response was 1.5 years (18.4 months [95% CI: 11.4-not estimable]). Data from the NP30179 study were recently published in the New England Journal of Medicine.
Columvi received its first worldwide approval in Canada in March 2023, and the European Medicines Agency’s Committee for Medicinal Products for Human Use recently granted a positive opinion recommending its approval in the European Union.
Glofitamab is under investigation in a randomized, open-label, multicenter Phase 3 study (STARGLO, NCT04408638) where patients with relapsed or refractory DLBCL receive glofitamab or rituximab in combination with gemcitabine + oxaliplatin (GemOx). Patients will receive up to 8 cycles of glofitamab IV or rituximab IV in combination with GemOx IV followed by up to 4 cycles of glofitamab monotherapy. The primary outcome measure is overall survival. The estimated study primary completion date is in April 2025.
Interested in data for other antibody therapeutics that have received marketing authorizations? Go to our searchable table of approved antibody therapeutics and those in regulatory review for more information.